利用高效液相色谱法测定甜茶叶中甜茶苷含量方法的研究(三)
发布时间:2021-07-28 19:14
编辑者:特邀作者夏德婷
3.5不同提取方式对甜茶苷含量测定的影响
所得结果如图4所示。先用石油醚浸泡处理的方式,虽然能去除色素、脂肪对测定结果的干扰,但是也导致少量甜茶苷损失、测定结果偏低。未经石油醚处理过的样品直接以30%乙腈为溶剂,以超声或高速均质的方式进行提取,所得的甜茶苷含量结果相对高。
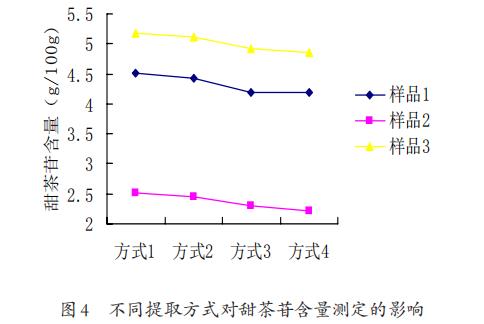
综合测定的液相色谱图,未经石油醚浸泡处理的样品测定液在色谱图中无干扰峰出现,因此样品不需要进行石油醚提前浸泡处理。
对比超声提取和高速均质提取,超声的设备成本较低,提取方式较为温和,因此选取超声提取作为样品前处理提取方式。
根据3.4和3.5的试验结果,甜茶叶中甜茶苷含量测定的前处理提取过程为:以30%乙腈为提取溶剂,以超声作为提取方式,提取时间为30min。
3.6回收率试验结果
结果如表3所示,本测定方法回收率测定结果为96.2%~102.3%,平均回收率为98.9%。
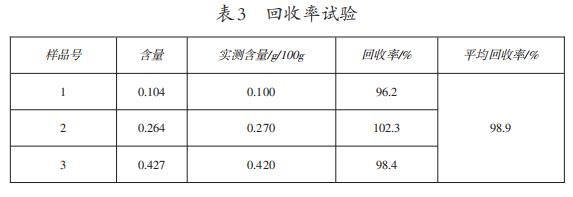
依据GB/T27404-2008《实验室质量控制规范食品理化检测》附录F对回收率的要求,被测组分含量在>100mg/kg时,回收率范围应在95%~105%。本测定方法符合要求,表明该方法回收率良好。
3.7精密度试验结果
6次重复测定所得结果及精密度结果见表4。
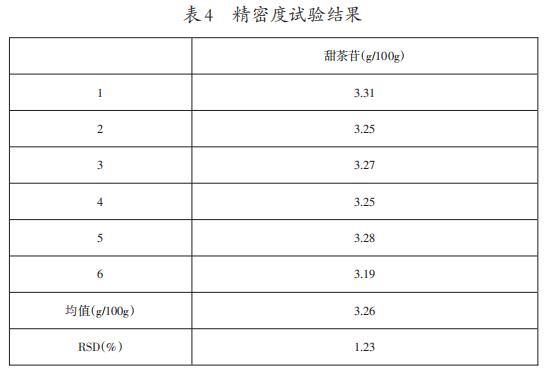
结果显示,使用此方法测定甜茶叶中的甜茶苷含量的精密度(RSD)符合GB/T27404-2008《实验室质量控制规范食品理化检测》附录F对精密度的要求(当被测组分含量在1%时,精密度应≤2.7%),因此,该方法的精密度良好。
4结论
本文建立了一种高效液相色谱法测定广西甜茶叶中甜茶苷含量的方法。经研究确定了甜茶叶测定液的制备流程:甜茶叶经30%乙腈溶液超声提取20min后,用30%乙腈定容至25mL,过滤,进样分析。色谱条件为:C18柱(250mm×4.6mm×5µm);流动相:0.05%磷酸溶液-乙腈(68+32);流速:1mL/min;检测波长:210nm;进样量:5µL;柱温:40℃;外标法定量。回收率和精密度试验结果表明,该方法精密度好,回收率好。因此,该方法具有简单、快捷、准确的优点,可以用于测定甜茶叶中甜茶苷含量。
声明:本文所用图片、文字来源《轻工科技》,版权归原作者所有。如涉及作品内容、版权等问题,请与本网联系删除。
相关链接:石油醚,甜茶苷,提取溶剂



















登录后才可以评论